医疗器械经营许可证办理分类及流程解析

医疗器械经营许可是指在中国境内从事医疗器械经营活动必须取得的行政许可。根据《医疗器械监督管理条例》及配套法规,其办理流程和要求因经营类别、企业类型及风险等级而异。本文将系统解析医疗器械经营许可证的主要分类及办理要点。
一、 核心分类:经营方式与经营范围
办理许可证时,首要明确的是企业的经营方式和拟经营的医疗器械范围。
- 按经营方式分类:
- 批发/销售: 将医疗器械销售给具有资质的经营企业或使用单位。
- 零售: 将医疗器械直接销售给消费者(通常指第二类医疗器械,部分第一类产品也可零售)。
- 为其他生产经营企业提供贮存、配送服务(第三方物流): 这是近年来明确并加强监管的一类,需单独申请资质。
- 按经营范围分类(基于产品风险等级):
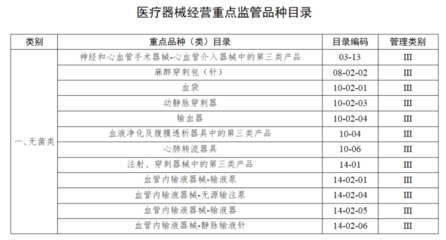
- 经营第二类医疗器械: 需要向所在地设区的市级药品监督管理部门申请办理《第二类医疗器械经营备案凭证》。这是较为普遍的许可类型,如血压计、体温计、医用口罩等。
- 经营第三类医疗器械: 需要向所在地设区的市级药品监督管理部门申请办理《医疗器械经营许可证》。这是监管最严格的类别,如植入式心脏起搏器、人工关节、血管支架等。
- 同时经营第二、三类医疗器械: 通常只需申请办理《医疗器械经营许可证》,其经营范围会涵盖允许经营的第三类及相应的第二类产品。
- 备注: 经营第一类医疗器械不需许可和备案,只需取得工商营业执照后按要求进行信息公示。
二、 特殊经营情形的分类管理
除上述基础分类外,针对特定经营模式,还有更细致的监管要求。
- 专门从事医疗器械第三方物流服务: 需参照经营第三类医疗器械的标准申请许可证,并特别审查其仓储、运输的软硬件条件及质量管理制度。
- 从事医疗器械批发兼零售业务: 需在申请时明确两种经营模式,其人员、场所、仓储、质量体系需同时满足批发和零售的要求。
- 经营需冷链管理的医疗器械: 无论是第二类还是第三类,对冷藏冷冻的仓储、运输设施设备及全程温度监控有额外严格要求。
- 为医疗器械注册人、备案人提供产品销售服务: 即“受托经营”,需在申请材料中明确委托关系,并承担相应的经营质量责任。
三、 办理流程概要与分类关联
办理流程的核心步骤一致,但具体细节因上述分类而异:
- 前期准备与自我评估: 根据拟经营的医疗器械类别(特别是第三类高风险产品名录)和经营方式,确定申请类型(是备案还是许可),并据此配置相应的人员(质量负责人、专业技术人员等)、经营与仓储场所、设施设备。
- 在线提交申请: 通过国家药品监督管理局的“医疗器械生产经营许可(备案)信息系统”或地方政务服务平台提交申请。关键是要准确选择“经营分类”和填写“经营范围”。
- 递交书面材料与现场核查:
- 第二类备案: 通常以形式审查为主,部分地区可能进行事后现场检查。
- 第三类许可及特殊情形: 必须经过药品监督管理部门的现场核查。核查重点与经营分类直接相关,例如:
- 经营无菌、植入、介入类第三类产品,对仓库洁净度、追溯系统要求极高。
- 从事零售,重点检查店面陈列、分区、药学服务人员资质。
- 从事批发和第三方物流,重点检查仓储面积、分区管理、计算机信息管理系统。
- 审批与发证: 现场核查通过后,监管部门作出准予许可或予以备案的决定,并发放相应的《医疗器械经营许可证》或《第二类医疗器械经营备案凭证》。
四、 重要提示
- “分类”是办理的起点: 企业在筹划之初就必须明确“卖什么(类别)”和“怎么卖(方式)”,这决定了全部的资源投入和申请路径。
- 法规动态更新: 医疗器械分类目录、经营监管办法等会适时调整,务必以申请时最新的法规和技术要求为准。
- 属地化管理: 具体办理细则、材料清单、办理时限可能因各省市药监局的要求有细微差异,申请前需详细咨询所在地监管部门。
医疗器械经营许可证的办理并非“一刀切”,而是基于产品风险和企业经营模式的一套精细化、分类别的管理体系。准确理解并定位自身的经营分类,是合法、高效取得资质,顺利开展业务的关键第一步。
如若转载,请注明出处:http://www.oqowcrcq.com/product/11.html
更新时间:2026-06-19 09:32:34